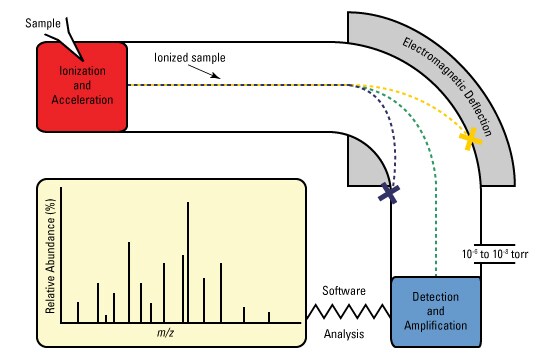
It is an analytcial chemistry technique that helps identify the amount and type of chemicals present in a sample by measuring the mass-to-charge ratio of ions.
No they are not. Tandem Mass Spectroscopy uses a tandem mass spectrometer. A Tandem Mass Spectrometer can be thought of as two mass spectrometers connected in series by a chamber that can break molecules into pieces.
Most of the times it is impractical to identify all the compounds and thus it is better to identify certain specific compunds which are of interest to our research. In a Tandem Mass Spectrometer, a sample is first sorted or weighed in the first Mass Spectrometer, it is then broken down into multiple pieces in the collision cell and then a piece or pieces are weighed and sorted in the second Mass Spectrometer
We took the following steps to identify the proteins:
The first thing that we though would make sense to do is to just read in the database of the available proteins(amino acid chains) and store them so that we can use them later to form tryptic peptides. For that we basically wrote a scraper fucntion that reads the file and scrapes all the proteins from it.
def readFasta(filename):
proteins = {}
inf = open(filename,'rU')
name = ""
dna = ""
for line in inf:
line = line.strip()
if line[0] == '>':
if name != "" :
proteins[name] = dna
name = line[1:]
dna = ""
else :
dna += line
proteins[name] = dna
So this basically gave us a dictionary of all the proteins in the file which can be easily accessible using the Accession.
FASTA : A text-based format for representing either nucleotide sequences or peptide sequences
A Tryptic peptide is a peptides that have been digested by Trypsin at sites [KR]|[^P].
Peptide is a shrot chain of amino acids linked by amide bonds
To convert the amino acid sequences into tryptic peptides, we wrote a regular expression based function that split the seqeuences at the Trypsin digestion sites.
def sep_peptides(peptide):
peptides = []
split_matches = re.finditer(r'[RK](?!P)', peptide)
prev_split_location = 0
for m in split_matches:
split_location = m.end()
peptides.append(peptide[prev_split_location:split_location])
prev_split_location = split_location
return peptide
Both the first 2 steps can be achieved by executing the following line of code either with ups.fasta (the small daatbase) or with UP000005640_9606.fasta (the big database) and the output will be saved in a regular .txt file
In [5]:
run read_fasta.py ups.fasta
To read and store the data for future purposes we wrote a parser function that stored the mass-intensity pairs of the spectrum along with the metadata like the mass to charge ratio, the scan number and the charge of the ion.
Mascot Generic Format(mgf) is a format in which each Mass Spectrometer readings is stored as a list of pairs of mass and intensity
`def read_mgf(fp): metadata = {} spectrums = [] with open(fp, 'r') as f: for line in f: line = line.strip() if line=="BEGIN IONS": spectrum = [] metadata = {} if line=="END IONS": spectrums.append( (metadata, spectrum) )
if line:
if line.count('=') == 1:
label, value = line.split('=')
if not label == 'CHARGE':
if '.' in value:
metadata[label] = float(value)
else:
metadata[label] = int(value)
else:
metadata[label] = value
elif line[0].isdigit():
mass, intensity = line.split(' ')
spectrum.append( (float(mass), float(intensity)) )
else:
pass
return spectrum`Having been already stored as metadata whilst parsing of the file, this one was a fairly easier step as it just required us to obtain the required numbers from the metadata.
Charge was already stored in the file and hence was accessible from the metadata
def calc_charge(metadata):
return int(metadata['CHARGE'][0]
Mass however wasn't already stored in the file and arithmetic operations were required to find out the mass
The formula used was (m+ 18.01)/z + 1.007 = PEPMASS(Stored in MGF File)
def calc_mass(metadata,charge):
pep_mass = metadata['PEPMASS']
mass = (pep_mass - 1.007)*charge - 18.01
return mass
The basic idea was that the mass of the peptide should match or be close to the mass of the spectrum. The accepted error was +/- 0.5 Daltons. The steps that we followed to achieve filtering by mass were as follows :
Dalton : 1/12 of Carbon-12 atom
To calculate the mass of the peptide we used the Amino Acid Mass Table. The only change we made to the table was to add 57.0219 to the mass of the Cytasine due to the changes made to it inside the Mass Spectrometer.
We just needed to get the list of all possible candidates which we achieved using:
def find_candidates(peptides,mass):
candidates = []
for peptide in peptides:
if abs(peptides[peptide] - mass) < 0.5 :
candidates.append(peptide)
return candidate
Y-ion mass = Amino Acid Mass + 19.018
A Simple Scoring Function :
A Better Scoring Function :
All the results can be generated running the following command
> python read_mgf.py <fasta_file> <mgf_file>
Scans Commercial Software found a result for : 282
Correct Scans With Simple Scoring Function and only Y-ions : 136
Correct Scans With Simple Scoring Function and both Y-ions and B-ions : 136
Correct Scans With Better Scoring Function and only Y-ions : 177
Correct Scans With Better Scoring Function and both Y-ions and B-ions : 178
Correct Scans with Better Scoring Function and only Y-ions : 125
Correct Scans with Better Scoring Function and both Y-ions and B-ions : 141
Possibly our scoring function isn't the most effective and it has problems dealing with a large database
In [ ]:
{kind=link}